Reporting of Methods Used to Ascertain Adverse Events of Special Interest (AESI) and Adverse Events Newly Signaled After Marketing Authorization of Drugs Approved Between 2018 and 2019
Abstract
Kyungwan Hong,1 Anisa Rowhani-Farid,1 Francis B. Palumbo,1 John H. Powers III,1,2 Linda Wastila,1 Peter Doshi1
Objective
Randomized clinical trials (RCTs) ascertain adverse events using 2 main approaches: (1) the nonsystematic approach collects data passively asking nontargeted questions relying on participants’ spontaneous responses, and (2) the systematic approach proactively collects data using standardized solicitation tools (eg, questionnaires).1 The ascertainment approach may affect the recorded rate of adverse events, affecting the interpretation of safety results from trials. Using publicly available trial documents, reported adverse event ascertainment methodologies used by regulatory agencies for marketing approval of new drugs were descriptively assessed.
Design
All new molecular entities and biologic license applications approved by the US Food and Drug Administration (FDA) in 2018 to 2019 were screened across a variety of indications. Where publicly available, clinical study reports, study protocols, and case report forms were obtained from trial registries, journals, and regulatory agencies’ websites. Reported ascertainment methods of 2 categories of adverse events were screened: adverse event of special interest (AESI) (adverse event types often predefined by investigators/sponsors to facilitate systematic assessment) and adverse events signaled by the European Medicines Agency Pharmacovigilance Risk Assessment Committee (EMA PRAC) within 12 months of market authorization (adverse-event types that conceivably could have been captured in preapproval trials). One researcher extracted the data. A second researcher verified a 10% random sample. Discrepancies were resolved through discussion.
Results
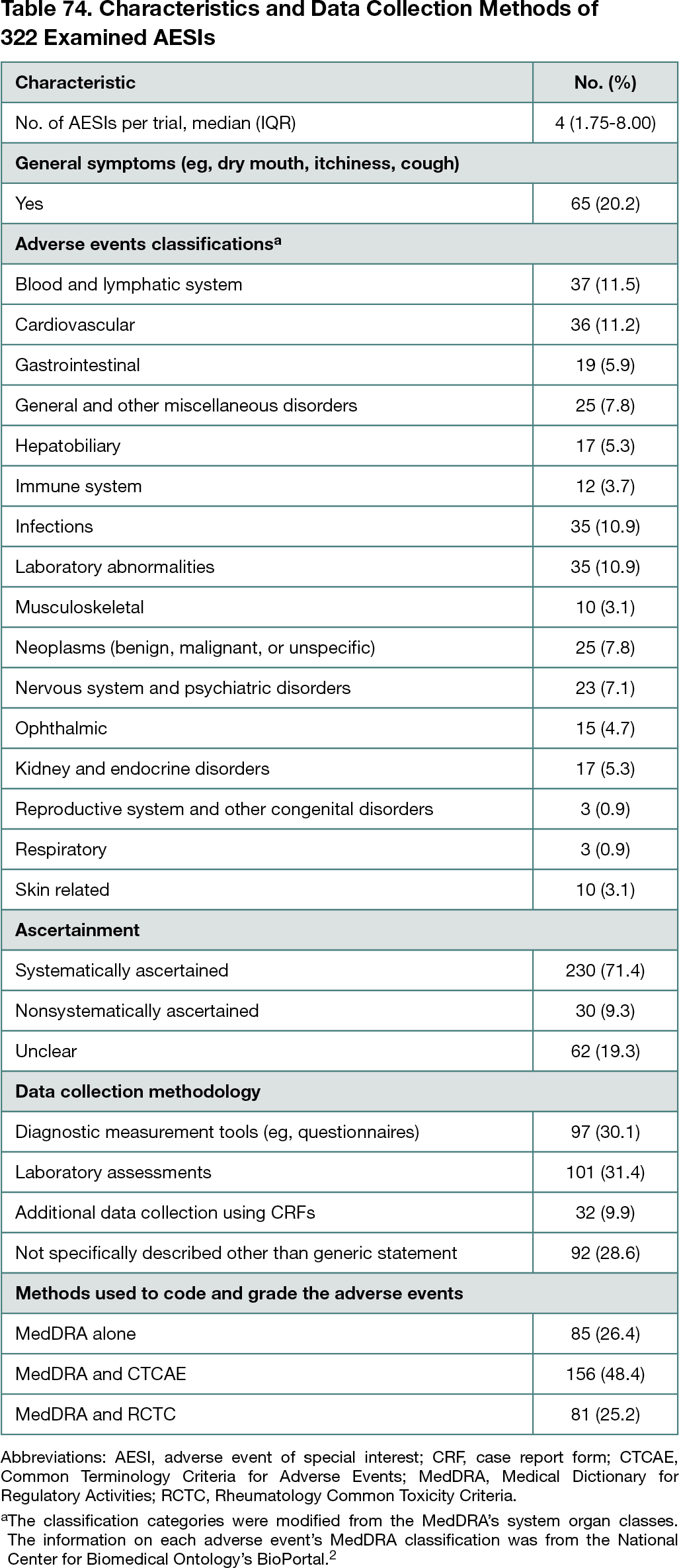
A total of 107 new molecular entities and biologic license applications approved in 2018 to 2019 were screened, and 31 drugs (29.0%) had publicly available underlying trial documents. Of 64 pivotal trials, AESIs were described in 54 clinical study reports (84.4%; 95% CI, 73%-92%), 33 study protocols (51.6%; 95% CI, 39%-64%), and 10 journal publications (15.6%; 95% CI, 8%-27%). A total of 322 AESIs were identified from trial documents. The median number of AESIs per trial was 4. Mainly using diagnostic measurement tools and laboratory assessments, 71.4% of AESIs (230 of 322; 95% CI, 66%-76%) were systematically ascertained, and 9.3% (30 of 322; 95% CI, 6%-13%) were nonsystematically ascertained. The ascertainment method of 19.3% (62 of 322; 95% CI, 15%-24%) was unclear (Table 74).2 In 38 of 54 trials (70.4%; 95% CI, 56%-82%), at least 1 AESI was nonsystematically ascertained and/or had unclear ascertainment methodology. From EMA PRAC reports, 1 newly signaled adverse event was identified and systematically ascertained using targeted questions in case report forms.
Conclusions
The ascertainment methodology for approximately 20% of AESIs was unable to be identified, even with access to underlying trial documents. Most systematically ascertained AESIs were laboratory abnormalities rather than direct measures of patient health. The lack of adequate reporting impedes the accurate interpretation of a drug’s adverse event profile. Given that regulatory agencies expect more rigorous data collection for AESIs,3 the finding that approximately 10% of AESIs were assessed nonsystematically suggests a need for more rigorous methodology to capture adverse events and increased regulatory oversight. Further investigation is needed to explore reporting of ascertainment methodologies used for other types of adverse events.
References
1. Mayo-Wilson E, Fusco N, Li T, et al. Harms are assessed inconsistently and reported inadequately, part 1: systematic adverse events. J Clin Epidemiol. 2019;113:20-27. doi:10.1016/j.jclinepi.2019.04.022
2. Whetzel PL, Noy NF, Shah NH, et al. BioPortal: enhanced functionality via new web services from the National Center for Biomedical Ontology to access and use ontologies in software applications. Nucleic Acids Res. 2011;39:W541-W545. doi:10.1093/nar/gkr469
3. US Food and Drug Administration. E19: optimisation of safety data collection. Accessed January 13, 2022. https://www.fda.gov/media/128313/download
1Department of Pharmaceutical Health Services Research, University of Maryland School of Pharmacy, Baltimore, MD, USA, hong.kyungwan@umaryland.edu; 2Department of Medicine, George Washington University School of Medicine, Washington, DC, USA
Conflict of Interest Disclosures
Kyungwan Hong reported receiving a financial assistance award from the US Food and Drug Administration (FDA) of the US Department of Health and Human Services (HHS) outside the submitted work. Kyungwan Hong, Anisa Rowhani-Farid, and Peter Doshi reported receiving salaries from the RIAT Support Center, funded by the Laura and John Arnold Foundation. John H. Powers III reported receiving consulting fees from Arrevus, Eicos, Eli Lilly, Evofem, Eyecheck, Fuji, Gilead, GlaxoSmithKline, Johnson & Johnson, Microbion, OPKO, Otsuka, Resolve, Romark, Shinogi, SpineBioPharma, UTIlity, and Vir outside the submitted work. Peter Doshi reported receiving travel funds from the European Respiratory Society and Uppsala Monitoring Center; grants from the FDA (through the University of Maryland and the Center of Excellence in Regulatory Science and Innovation), the Laura and John Arnold Foundation, the American Association of Colleges of Pharmacy, the Patient-Centered Outcomes Research Institute, the Cochrane Methods Innovations Fund, and the UK National Institute for Health Research; reported being an unpaid Innovation in Medical Evidence Development and Surveillance steering committee member at the Reagan-Udall Foundation for the FDA; and being an editor at The BMJ. No other disclosures were reported.
Additional Information
Peter Doshi is a co–corresponding author.